Rokkarin asenteella syöpää vastaan
 Tämä on kertomus tietojenkäsittelijänä aloittaneesta ja sittemmin bioinformatiikan maisteriohjelmaan jämähtäneestä ikuisuusopiskelijasta, Riku Kataisesta, joka tarttui sattumalta itseään suurempaan haasteeseen. Alkujaan Veli Mäkisen SuDS-ryhmässä Kumpulassa tutkijanuransa aloittanut hevi-klišee loikkasi Meilahteen Lauri Aaltosen syöpägenetiikan tutkimusryhmään. Pääasiallisena toimenkuvana oli laatia visualisointi- ja analyysiohjelma geneettiselle sekvenssidatalle, jolla olisi mahdollista löytää geenivirheitä syöpäpotilaista. Vaikka ohjelman luominen ylitti alkuvaiheissa haastavuudessaan kaikki sietokyvyn rajat, ei vaivannäkö mennyt hukkaan. Antaa Rikun kertoa, mitä oikein tapahtui.
Tämä on kertomus tietojenkäsittelijänä aloittaneesta ja sittemmin bioinformatiikan maisteriohjelmaan jämähtäneestä ikuisuusopiskelijasta, Riku Kataisesta, joka tarttui sattumalta itseään suurempaan haasteeseen. Alkujaan Veli Mäkisen SuDS-ryhmässä Kumpulassa tutkijanuransa aloittanut hevi-klišee loikkasi Meilahteen Lauri Aaltosen syöpägenetiikan tutkimusryhmään. Pääasiallisena toimenkuvana oli laatia visualisointi- ja analyysiohjelma geneettiselle sekvenssidatalle, jolla olisi mahdollista löytää geenivirheitä syöpäpotilaista. Vaikka ohjelman luominen ylitti alkuvaiheissa haastavuudessaan kaikki sietokyvyn rajat, ei vaivannäkö mennyt hukkaan. Antaa Rikun kertoa, mitä oikein tapahtui.
Sattuman ohjaamana
 Noin kaksi vuotta sitten ryhdyin SuDS-ryhmässä ohjelmoimaan Javalla kevyttä käyttöliittymää, jolla oli tarkoitus visualisoida ryhmämme algoritmin toimintaa koskien geneettistä dataa. Tuotos sattumalta muistutti järjestelmää, jollaista Aaltosen ryhmässä oltiin kaivattu ja jatkoin täten kehitystä biologisempaan suuntaan. Huomasin kuitenkin pian, että tarvitsen tutkijoita kertomaan minulle, mitä ominaisuuksia he tarvitsevat ja millä tavalla niiden tulisi toimia. Siirryin Biomedicumiin nykyisen ryhmäni pariin työstämään ohjelmaa, joka mahdollisti jatkuvan vuorovaikutuksen ohjelman loppukäyttäjien kanssa.
Noin kaksi vuotta sitten ryhdyin SuDS-ryhmässä ohjelmoimaan Javalla kevyttä käyttöliittymää, jolla oli tarkoitus visualisoida ryhmämme algoritmin toimintaa koskien geneettistä dataa. Tuotos sattumalta muistutti järjestelmää, jollaista Aaltosen ryhmässä oltiin kaivattu ja jatkoin täten kehitystä biologisempaan suuntaan. Huomasin kuitenkin pian, että tarvitsen tutkijoita kertomaan minulle, mitä ominaisuuksia he tarvitsevat ja millä tavalla niiden tulisi toimia. Siirryin Biomedicumiin nykyisen ryhmäni pariin työstämään ohjelmaa, joka mahdollisti jatkuvan vuorovaikutuksen ohjelman loppukäyttäjien kanssa.
Aluksi ajattelin, että tämähän on täydellinen tilanne ohjelman kehittäjälle, mutta kävi hyvin nopeasti ilmi, että olin tunkenut lusikkani supermassiiviseen soppakattilaan. Ei riittänyt, että biologisen datan käsittely vilisi poikkeustapauksia, eikä se, että käyttöliittymän suunnittelu, toteutus ja testaus vaatisi vähintään keskikokoisen työryhmän, vaan näiden lisäksi datamäärät kymmenenkertaistuivat heti siirtymiseni jälkeen useisiin gigatavuihin per näyte. Olin päättänyt, että ohjelmalla olisi mahdollista visualisoida useita näytteitä samanaikaisesti ja sen tulisi vielä toimia nopeasti tavallisella pöytäkoneella ja yhdellä gigalla muistia, joten nerokkaille koodi-inspiraatioille oli suuri tarve. Onneksi uusi ryhmä oli täynnä inspiroivaa, älykästä ja iloista väkeä, joiden parissa luomistyön vaikeudet jaksoi kantaa vaikka vasemmalla kädellä, eikä inspiraatioitakaan tarvinnut kauaa odottaa.
Mihin oikein ryhdyinkään...
Ohjelmointikokemusta olin kartuttanut Velin ryhmässä noin kaksi vuotta Java-kurssien lisäksi, joten aivan tyhjän päältä ei tarvinnut ponnistaa. Bioinformatiikan maisteriohjelmassa opitut asiat olivat myös erittäin tärkeässä asemassa, jotta ymmärtäisin tutkijoiden tarpeet ja taustalla piilevät biologiset ongelmat.
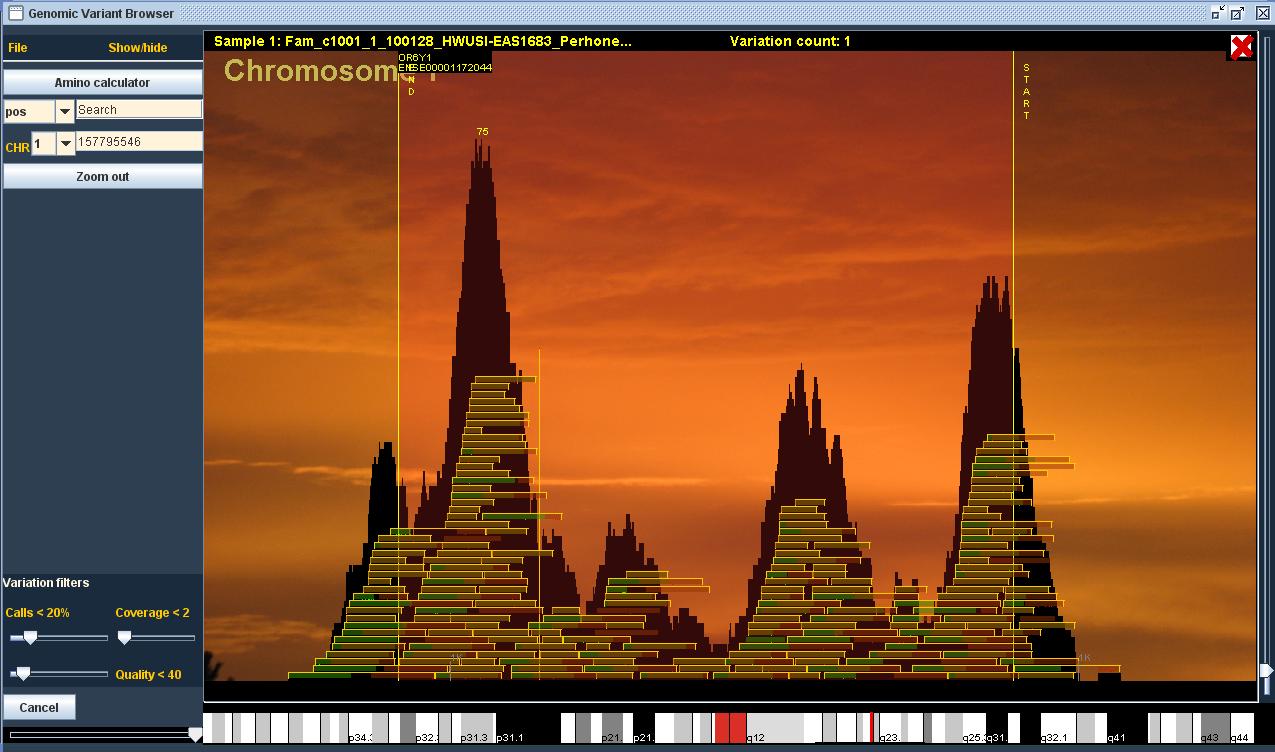
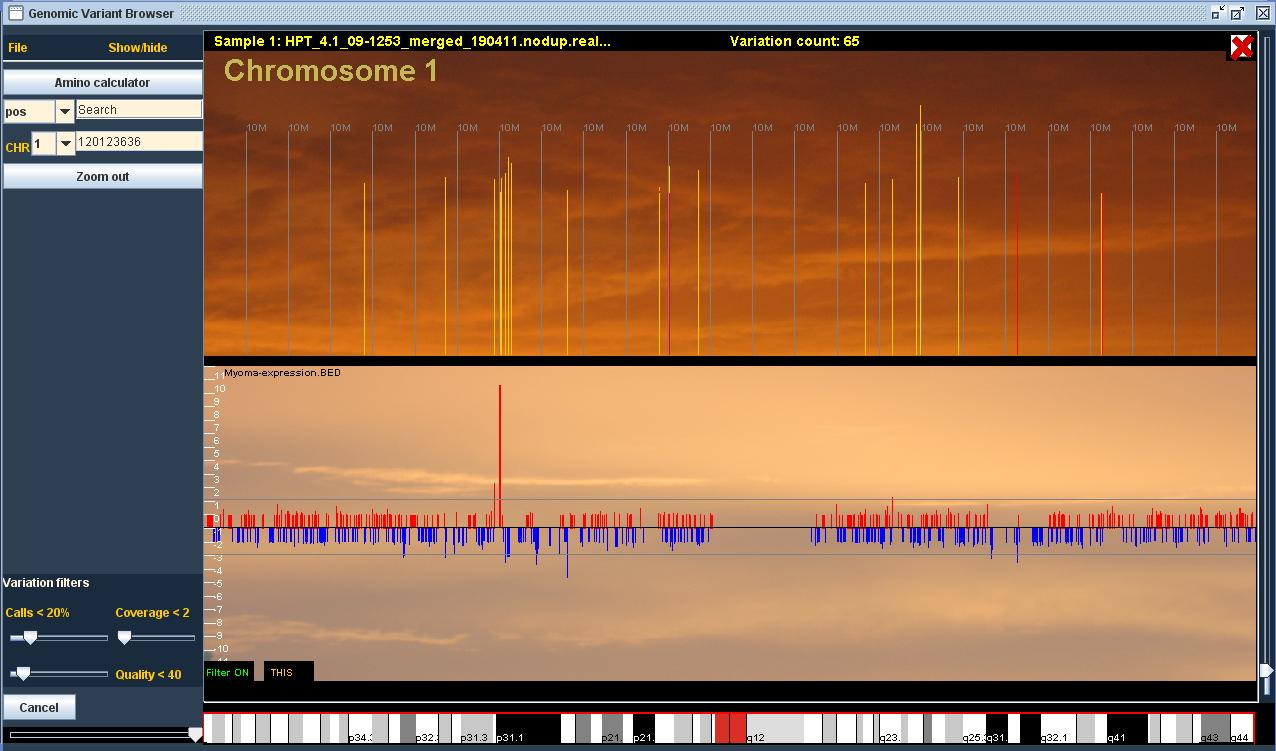 Halusin ensisijaisesti luoda käyttäjälle intuitiivisen, selkeän ja ennen kaikkea nopeasti reagoivan käyttöliittymän, jotta tutkijan flow ei karkaisi liiallisen odottelun vuoksi. Päätökseni tehdä ohjelma läppärilläkin toimivaksi asetti rajoitukset muistinkäytön ja laskentatehon suhteen. Näiden datamäärien kanssa oli selvää, että muistiin ei voinut ladata kuin murto-osa koko datasta, joten tiedostohaut tuli toteuttaa erittäin nopeaksi reaaliaikaisuuden illuusion ylläpitämiseksi. Käytännössä käyttäjän zoomatessa kromosomi- tasolta lähemmäksi sekvenssiä, ohjelma hakee ainoastaan tarpeellisen (ruudulla näkyvän) osan tiedostoista muistiin. Esimerkiksi kaikista lähin zoomaus näyttää ruudun alalaidassa DNA-sekvenssin, jonka haen 3GB:n genomitiedostosta käyttäjän liikkeiden mukaan. Parhaimmillaan ohjelma hakee samanaikaisesti tietoa geeni-annotaatio-, DNA-sekvenssi-, variaatio- ja readitiedostoista ja piirtää kaiken tiedon ruudulle. Tiedostomäärä kasvaa tietenkin käyttäjän avatessa useampia näytteitä. Esimerkiksi sadalla näytteellä ohjelman tulee käsitellä yli 500GB datamäärää.
Halusin ensisijaisesti luoda käyttäjälle intuitiivisen, selkeän ja ennen kaikkea nopeasti reagoivan käyttöliittymän, jotta tutkijan flow ei karkaisi liiallisen odottelun vuoksi. Päätökseni tehdä ohjelma läppärilläkin toimivaksi asetti rajoitukset muistinkäytön ja laskentatehon suhteen. Näiden datamäärien kanssa oli selvää, että muistiin ei voinut ladata kuin murto-osa koko datasta, joten tiedostohaut tuli toteuttaa erittäin nopeaksi reaaliaikaisuuden illuusion ylläpitämiseksi. Käytännössä käyttäjän zoomatessa kromosomi- tasolta lähemmäksi sekvenssiä, ohjelma hakee ainoastaan tarpeellisen (ruudulla näkyvän) osan tiedostoista muistiin. Esimerkiksi kaikista lähin zoomaus näyttää ruudun alalaidassa DNA-sekvenssin, jonka haen 3GB:n genomitiedostosta käyttäjän liikkeiden mukaan. Parhaimmillaan ohjelma hakee samanaikaisesti tietoa geeni-annotaatio-, DNA-sekvenssi-, variaatio- ja readitiedostoista ja piirtää kaiken tiedon ruudulle. Tiedostomäärä kasvaa tietenkin käyttäjän avatessa useampia näytteitä. Esimerkiksi sadalla näytteellä ohjelman tulee käsitellä yli 500GB datamäärää.
Mitä ohjelmalla tehdään?
Ohjelman pääasiallinen tarkoitus on geenivirheiden etsiminen sairastuneiden ihmisten genomista. Tätä varten ohjelma tarjoaa välineet usean (Suurimmassa datasetissämme on 86 näytettä) potilaan yhtäaikaiseen vertailuun. Ohjelma laskee variaatioiden aiheuttamat aminohappomuutokset ja mahdollistaa erilaisten filtterien, sekä kontrollien käytön. Ohjelmaan voi lisätä myös BED-tiedostoja, joihin voi sisällyttää omia kiinnostavia alueita tai vaikka ekspressiodataa.
Mistä näytteet ohjelmaan?
Ohjelmalla avataan potilaiden DNA-sekvenssidataa, joka on eristetty verestä tai kudosnäytteestä. DNA-sekvenssi luetaan analyysilaitteella pienissä pätkissä ja tuloksena saadaa tiedosto, jossa on miljoonia sekvenssipätkiä/readeja pituudeltaan noin 50-150 emästä. Tämän jälkeen readit linjataan ihmisen referenssigenomiin, joka kertoo eroavaisuudet (mahd. mutaatiot) potilaan ja referenssin välillä. Ohjelmalla avataan tiedosto, joka sisältää tarvittavat tiedot variaatioista ja readeista.
Intensiivisen kynsienpurennan, hiustenrevinnän ja kiroilun lomassa onnistuin viemään projektia hiljalleen eteenpäin ja suurimmat inspiraatiot syntyivät, omituista kyllä, luovassa maanantaikohmelossa. Saatoin saada villin idean, jota seurasi viiden tunnin taukoamaton naputtelu. Ympärillä oleva hälinä ei rekisteröitynyt aivoihin, kunnes koodin toimivuuden huomattuani nostin käteni pystyyn ja huusin voimasanoin ilon parahduksia ja tarpeetonta itseylistystä. Tällaisia lähes uskonnollisia hetkiä olen kokenut noin kolme ja ne ovat mahdollistaneet koko ohjelman olemassaolon tänä päivänä.
Mitä nyt?
Nyt ohjelmani, työnimeltään RikuRator, on tutkimuskäytössä ja se on näyttänyt voimansa geenivirheiden löytämisessä. Seuraava etappi onkin ohjelman virallinen julkaisu, joka tapahtuu toivottavasti vielä tänä syksynä. Olen matkan varrella oppinut Aaltosen ryhmässä paljon syöpägenetiikan tutkimuksesta ja saankin piakkoin oman kohteeni kilpirauhaskasvaimiin liittyen, josta toivottavasti riittää materiaalia aina väitöskirjaan asti. Olen ollut onnekas saadessani toteuttaa omaa kunnianhimoani ja luovuuttani Velin ja Laurin ryhmissä, vaikka olen itse välillä ollut skeptinen oman visioni toteutusmahdollisuuksista. Usko ja kärsivällisyys on nyt hetkellisesti palkittu, mutta saapahan nähdä, mihin sfääreihin seuraava teknologinen askel Riku-paran vie.
RikuRator
RikuRatorin esittelyvideo Youtubessa (suosittelemme 720p tarkkuutta ja uuden ikkunan avaamista)
Luotu
07.09.2011 - 12:42
 Laitoksen professori Petri Myllymäki on valittu Helsingin yliopiston ja Aalto-yliopiston yhteisen tietotekniikan tutkimuslaitoksen (HIIT – Helsinki Institute for Information Technology) johtajaksi 1.8.2015 alkavaksi viisivuotiskaudeksi. Petri seuraa tehtävässä Aalto-yliopiston professori Sami Kaskea. Koska HIITin johtajan tehtävä on kokoaikainen, vapautetaan Petri (PM) samaksi ajaksi omasta professuuristaan, ja laitoksen johtajan (JP) suorittama työstävapautushaastattelu on siten paikallaan.
Laitoksen professori Petri Myllymäki on valittu Helsingin yliopiston ja Aalto-yliopiston yhteisen tietotekniikan tutkimuslaitoksen (HIIT – Helsinki Institute for Information Technology) johtajaksi 1.8.2015 alkavaksi viisivuotiskaudeksi. Petri seuraa tehtävässä Aalto-yliopiston professori Sami Kaskea. Koska HIITin johtajan tehtävä on kokoaikainen, vapautetaan Petri (PM) samaksi ajaksi omasta professuuristaan, ja laitoksen johtajan (JP) suorittama työstävapautushaastattelu on siten paikallaan.