Rocking against cancer
 This is the story of Riku Katainen, an eternal student who started with computer science and then got stuck in the bioinformatics Master’s programme. By chance, he grappled a challenge larger than himself.This heavy-metal cliché defected from his original research career in Veli Mäkinen’s SuDS group in Kumpula to Lauri Aaltonen’s research group on cancer genetics in Meilahti.His primary duties involve creating visualisation and analysis software for genetic sequence data, with which to discover gene defects in cancer patients.Though the challenge of creating the program initially brought Riku to the limits of his tolerance, all his hard work bore fruit in the end.Let us hear him tell the story.
This is the story of Riku Katainen, an eternal student who started with computer science and then got stuck in the bioinformatics Master’s programme. By chance, he grappled a challenge larger than himself.This heavy-metal cliché defected from his original research career in Veli Mäkinen’s SuDS group in Kumpula to Lauri Aaltonen’s research group on cancer genetics in Meilahti.His primary duties involve creating visualisation and analysis software for genetic sequence data, with which to discover gene defects in cancer patients.Though the challenge of creating the program initially brought Riku to the limits of his tolerance, all his hard work bore fruit in the end.Let us hear him tell the story.
Guided by chance
About two years ago, in the SuDS group, I started programming a light user interface with Java for visualising the group’s algorithm operating on genetic data. The result happened to resemble a system that Aaltonen's group had wished for, and so I continued to develop it in a more biological direction. However, I soon noticed that I would need researchers to tell me what features they would need and how they should work. I transferred to Biomedicum and my current team to work on the program, which enabled me to be in constant interaction with the end users.
At first I thought this was the ideal situation for a software developer, but it soon turned out that I had opened a super-massive can of worms.It wasn’t enough that the biological data was full of exceptions, or that the design, implementation and testing of the user interface would require a medium-sized work team, but the amounts of data also increased exponentially to several gigabytes per sample soon after I transferred.I'd decided that the program should be able to visualise several samples at the same time, and it should work quickly on an ordinary PC with one GB of memory, so I had a great need for some brilliant code inspiration. Luckily, the new team was full of inspiring, intelligent and happy people, whose company helped me carry the creative burden, and I didn’t even need to wait long to be inspired.
What did I get myself into…
I had developed my programming experience in Veli’s team for about two years, in addition to the Java courses, so I did have a solid base to start from. The things I’d learned in the MBI were also a great help in understanding what the researchers needed and the biological problems in the background.
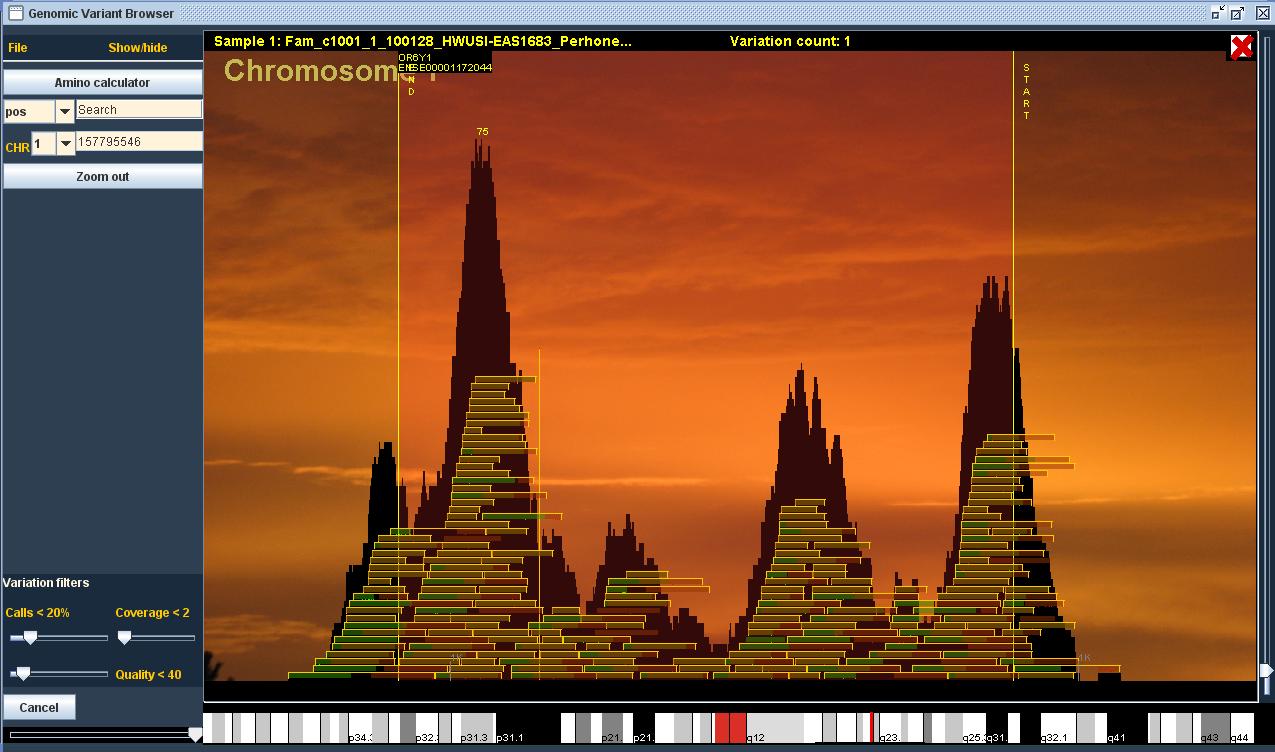
Primarily, I wanted to create an intuitive, plain and, most of all, quicly reacting user interface, so that the researcher’s flow wouldn’t be interrupted by long waits. My decision to make the program work on laptops, too, set its own restrictions on memory use and computing capacity. With these amounts of data, it was clear that it wouldn’t be possible to load more than a fraction of the whole data into the memory, so file searches had to be made very fast in order to keep up the illusion of real-time action. In practice, as the user zooms in from the chromosome level to the sequence, the program only finds the necessary (on-screen) part of the files. The most detailed zoom, for instance, shows at the bottom of the screen the DNA sequence that I can find from the 3GB genome file according to the user’s movements. At best, the program finds information from the gene-annotation, DNA-sequence, variation and read files simultaneously, and draws all the information on-screen. The number of files naturally increases as the user opens more samples. With a hundred samples, for example, the program must handle over 500GB of data.
What does the program do?
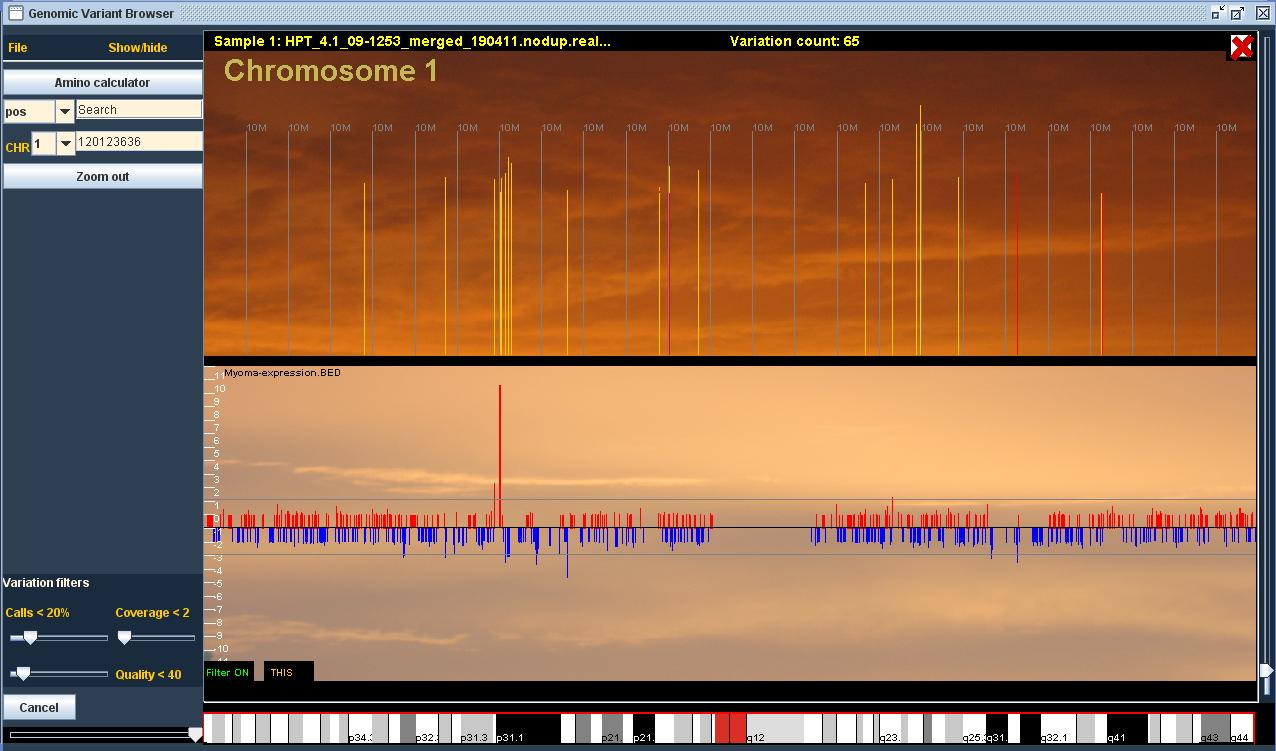
The main purpose of the program is to find gene defects in the genome of people with diseases. For this purpose, the program offers the tools to compare several patients at once (our largest data set contains 86 samples). The program calculates the amino-acid fluxes caused by variation, and enables the use of various filters and controls. You can also add BED files to the program to include the areas you are interested in, such as expression data.
Where do the samples come from?
The program is used to open up patients’ DNA sequence data that has been extracted from blood or tissue samples. The DNA sequence is read in short sequences with an analysis apparatus, which results in a file with millions of short sequences/ reads that are around 50-150 base pairs long. After this, the reads are lined up with the human reference genome to show possible differences (mutations) between the patient and the reference. The program can be used to open a file with necessary information on variations and reads.
During intensive nail-biting, hair-pulling and swearing, I managed to bring the project forwards slowly, and funnily enough, the greatest inspirations came during my creative Monday hangovers. I could have a wild idea followed by five hours of unbroken tapping on the keyboard. The noises around me did not register in my brain, until I lifted up my hands after realising the code worked, and broke out in four-letter words of happiness and unnecessary self-praise. I’ve had about three of these almost religious experiences and they have made the whole program possible today.
What now?
Now my program, called the RikuRator, is used in research and it has shown its strength in finding gene defects.The next phase is the official publication of my program, which will hopefully happen this autumn.During this journey, I have learned a lot about cancer genetics research in Aaltonen’s group, and soon I will have my own topic in the study of thyroid tumours, where I hope to find enough material for a doctoral thesis.I’ve been lucky in having the opportunity to realise my own ambitions and creativity in Veli’s and Lauri’s groups, though I’ve sometimes been sceptical of the possibility to realise my vision.My faith and patience have now been rewarded momentarily, but we’ll see to which heavenly spheres the next technological step will take poor Riku.
RikuRator
A presentation of the RikuRator on Youtube (we recommend a resolution of 720p tarkkuutta and opening a new window)
Created date
09.09.2011 - 17:47
